Averaging with DENSS
DENSS reconstructs particles in an iterative fashion which begins by filling the entire grid of points with random values. As a result of this, each run of DENSS with identical input parameters yields a slightly different result. These results, while different at high resolution, should be similar at low resolution. Therefore, to determine the low-resolution density, one must run DENSS multiple times and average the results.
There are currently two approaches to averaging maps made easy with DENSS. The original approach uses EMAN2, which is a large suite of scientific image processing software most often used for electron microscopy. It’s very high quality and well supported, however it requires installation of EMAN2 (but that’s pretty easy now). There have been reports from some DENSS users about issues getting EMAN2 to run on Windows. Also, EMAN2 is quite large, and DENSS only needs one small part of it, and most of the package goes unused by DENSS users. So, we decided to write our own alignment and averaging procedure built-in to DENSS. In our tests it works on Windows as well. If you would like to perform the averaging procedure using EMAN2, click here. Note that we no longer maintain the EMAN2 averaging procedure (though it should still work). Both the EMAN2 and the built-in averaging procedures are parallelized for taking advantage of multi-core machines.
The primary script for performing multiple runs of DENSS and averaging the results is denss.all.py. To run with default parameters, type:
> denss.all.py -f 6lyz.outThe default will run 20 reconstructions of DENSS, perform enantiomer generation and selection, alignment, averaging, and resolution estimation. By default, this will only run on a single core. To enable parallelization, invoke the -j option with the number of cores. For example, to run using 4 cores in parallel with default parameters, type:
> denss.all.py -f 6lyz.out -j 4On four cores, the full procedure will take around 2-3 hours on a modern system.
If this is the first time you have run denss.all.py using this input file, this will create a folder called “6lyz” containing the results. If you have run this command before with the same input file or that directory name already exists, denss.all.py will create a new directory and append incrementing numbers to the end of the directory name so that your previous results are not overwritten. If you would like to change the output filename prefix, set the -o option. If you would like to enter additional options for running the individual DENSS reconstructions, you can give them directly to denss.all.py, just as you would for a single reconstruction with denss.py. For example, to force DENSS to use a different Dmax than in the GNOM file, type:
> denss.all.py -f 6lyz.out -j 4 -d 55.0If you would like to run more reconstructions than the default 20, you can invoke the –nmaps option as follows:
> denss.all.py -f 6lyz.out -j 4 -d 55.0 --nmaps 100Enantiomer Generation and Selection
By default, denss.all.py will attempt to deal with possible enantiomer ambiguity. Enantiomers (i.e. mirror images of particles) are ambiguous in solution scattering and yield identical scattering profiles. It is possible for DENSS to generate results with different enantiomers based on the random seed that it starts with. Because of this, different enantiomers may get averaged together if not accounted for. For some particle shapes, this is not so much of an issue because the possible enantiomers may only be distinguishable at resolutions higher than can be reconstructed. However for more complex particle shapes, different enantiomers may be clearly distinguishable and yield incorrect results if averaged together. denss.all.py will generate and compare each enantiomer to the reference volume to select which enantiomer agrees best. Then the averaging procedure will continue as normal using only the best enantiomers. To disable enantiomer selection, invoke the -en_off option.
Here we will take a look at a particularly complex particle shape, SusF (PDB ID 4FE9). This protein was selected from the PDB as exhibiting the single highest ambiguity score as estimated by AMBIMETER (a score of 3.019). The complexity of the shape makes incorrect enantiomer selection a serious problem. It is also a good demonstration of the ability of DENSS to reconstruct complex shapes. Note the image below on the left shows a single reconstruction of denss.py that created the correct enantiomer. Shown in the middle is an example of a single reconstruction that selected the incorrect enantiomer. Shown on the right is the final reconstruction after generating and selecting for the correct enantiomers.



It should be noted that this procedure does not ensure that the final averaged reconstruction represents the actual correct enantiomer, it just ensures that all of the reconstructions used in the averaging procedure have the same handedness. There is no way to to select the correct enantiomer without some ancillary data, since enantiomers are ambiguous in solution scattering.
Output
The output for the reconstructions and averaging will be stored in output folder. A log file for each reconstruction and for the averaging will be included. The main log file containing statistics about averaging will be called “6lyz_final.log”. The averaging statistics include correlation scores for each map to the reference. Maps will be filtered to remove outliers (>2 standard deviations from the mean correlation score), and any maps failing this test will be marked with an “F”. The mean and standard deviation of correlation scores will also be given. These values can be used to estimate how reproducible the reconstructions are. A small standard deviation suggests the reconstructions are relatively unique, whereas a large standard deviation suggests there is significant variation. Note that the absolute value of the correlation scores is dependent on the size of the grid and the volume of the grid that the particle occupies, so relating one system to another is not always appropriate.
The final averaged reconstruction will be called “6lyz_avg.mrc”. Each reconstruction after enantiomer selection and alignment will be saved as “6lyz_?_aligned.mrc” where the “?” refers to the reconstruction number.
Resolution
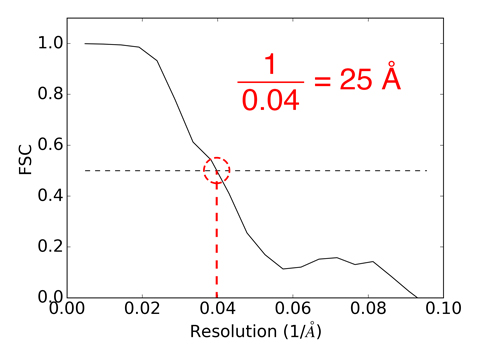
The averaging procedure works by aligning each reconstruction against a reference map. The reference map is calculated using a binary tree algorithm, where several pairs of maps are aligned and averaged in steps, ultimately generating a simple average. The Fourier Shell Correlation comparing each reconstruction to the reference is calculated, and the average of all FSC curves is calculated and saved in a file named “6lyz_fsc.dat”. This plot can be used to estimate resolution where the FSC curve falls below 0.5. Take the reciprocal of that x-axis value, and that is your estimated resolution in Å. For convenience this resolution is estimated and printed to the log file and screen for you. If the python module matplotlib is available, you can use the supplied fsc2res.py script to make a plot of the FSC and estimated resolution which will be saved to a png file. To estimate resolution by comparing with a known structure see denss.align.py and denss.calcfsc.py below and the Tips page.
Helpful Scripts
In addition to denss.all.py, there are several helper scripts to perform various tasks that may be useful to have exposed separately:
denss.align.py – A tool for aligning electron density maps. This script can be used if you would like to align an electron density map (or several maps) you have generated to another electron density map or to an atomic model. denss.align.py supports enantiomer selection as well. The reference will be an electron density map, either the given .mrc file, or a map calculated from a PDB model if a .pdb file. For example, to align the 6lyz_average.mrc map we created above to the 6lyz.pdb file, simply type:
> denss.align.py -f 6lyz_average.mrc -ref 6lyz.pdbThis will save the aligned map as “6lyz_average_aligned.mrc” and save a log file with useful alignment statistics as “6lyz_average_aligned.log”.
denss.align2xyz.py – A tool for aligning an electron density map such that its principal axes of inertia are aligned with the x,y,z axes.
denss.align_by_principal_axes.py – A tool for aligning an electron density map to another electron density map based only on alignment of principal axes (no minimization).
denss.average.py – A tool for averaging multiple pre-aligned electron density maps. In some cases, you may want to average a selection of maps that are pre-aligned. This script performs this simple task for you.
denss.align_and_average.py – A tool for aligning and averaging multiple electron density maps. This script is essentially everything that denss.all.py is, except does not perform all the individual reconstructions of denss.py. denss.align_and_average.py will take set of maps as a space separated list (which on many terminals can be simplified with wildcard characters), and perform enantiomer selection, alignment and averaging. This can be helpful if you have already calculated maps and simply want to average them. For example, to average the 20 reconstructions of 6lyz, you could type:
> denss.align_and_average.py -f 6lyz_*[0-9].mrcHere we have used bash wildcard expansion (the “*[0-9]”) to tell the shell to select all files that start with “6lyz_” and end with a number followed by “.mrc” to select the maps generated by denss.py. Note that in this case we did not simply use “6lyz_*.mrc” because this would also select all of the support maps saved by denss.py. This script can also be helpful if you would like to simply take a manual selection of maps and average them. For example, if you have invoked NCS averaging in denss.all.py, its possible that some of the reconstructions selected the wrong axis of symmetry. In such cases you would want to just select the maps with the correct axis of symmetry and average those separately from the rest. After manually inspecting the maps, you could run denss.align_and_average.py and give a space separated list of each correct map, or it might be easier just to copy the reconstructions with the correct symmetry axis into a new folder, and run denss.align_and_average.py in the new folder.
denss.refine.py – A tool for refining an electron density map from solution scattering data. One of the downsides of averaging is that the final averaged map is unlikely to have a corresponding scattering profile that matches the experimental profile, since it is an average of many different maps. To generate a map that has the benefits of averaging and having a scattering profile matching the data, you can use the denss.refine.py script. This script runs exactly like denss.py, with the added ability to accept an electron density map to start with (rather than the random electron density map that denss.py starts with), akin to refining the averaged map against the data. For example, to refine the averaged map for the 6lyz case above, simply type:
> denss.refine.py -f 6lyz.out -rho 6lyz_average.mrcdenss.calcfsc.py – A tool for calculating the Fourier Shell Correlation between two pre-aligned MRC formatted electron density maps.
denss.get_info.py – Print some basic information about an MRC file. Prints the grid shape (i.e. the number of grid points in each dimension), the size of the box in Å, and the voxel size in Å, as well as various useful statistics about the map.
> denss.get_info.py -f 6lyz.mrc
Grid size: 32 x 32 x 32
Side length: 145.222504 x 145.222504 x 145.222504
Voxel size: 4.538203 x 4.538203 x 4.538203
Voxel volume: 93.465606
Total number of electrons: 10000.000723
Min/max density: -0.000475 , 2.403097
Center of mass in angstroms: [ 0.541977 -1.644859 2.004769 ]
Mean Density (all voxels): 0.00327
Std. Dev. of Density (all voxels): 0.06418
RMSD of Density (all voxels): 0.06426
Modified Mean Density (voxels >0.01*max): 0.66008
Modified Std. Dev. of Density (voxels >0.01*max): 0.62799
Modified RMSD of Density (voxels >0.01*max): 0.91108denss.pdb2mrc.py – A tool for calculating electron density maps from pdb files. The map will be calculated using a real space form factor equation using the commonly used Cromer-Mann Gaussian summation with additional terms for excluded solvent and the hydration shell. Low-resolution maps can also be calculated using the –resolution option, which applies a Gaussian blur to the output map (but does not affect the scattering profiles calculation). You can also provide an experimental scattering profile to fit the bulk solvent density and hydration shell contrast parameters to best match the data.
denss.mrc2sas.py – A tool for calculating scattering profiles from MRC formatted electron density maps.
denss.regrid.py – A tool for regridding a 1D scattering profile from one set a q values to another set of q values using interpolation.
denss.mrcops.py – A tool for performing basic operations on MRC formatted electron density maps. This script allows you to resample or reshape a map. To resample a map, invoke the -v option to change the voxel size. For example, say you have a map that has a current grid size of 32x32x32 and you want to double the sampling to 64x64x64. First use the denss.get_info.py script to determine what the current voxel size is, which will print something to the screen like in the above example.
Then calculate what the new voxel size should be (4.538203 * 32 / 64 = 2.2691015) and use the -v option of denss.mrcops.py to resample the map:
> denss.mrcops.py -f 6lyz.mrc -v 2.2691015This will save a new map called 6lyz_resampled.mrc. You can also change the size of the box without resampling, which will simply pad the map with zeros or crop the map at the edges, using either the -n or –side options. Additionally, you can rescale the density in the map to have a specified number of electrons (useful for absolute scaling in e-/Å3), or set a minimum threshold for the map (where lesser values will be set to zero), or to generate the enantiomer (–zflip option).