Basic Usage
Here we will use the provided 6lyz.out file for this tutorial, created by GNOM. We will also show how to use the 6lyz.dat file (in case a GNOM formatted .out file is not available for your data). DENSS accepts a variety of file types, including 3-column ASCII text .dat files containing raw data or a smooth fit (columns q, I, error), 4-column ASCII text .fit files containing raw data and the smooth fit (columns q, I, error, fit), and GNOM .out files.
First, to simply run DENSS with default parameters (suitable for many cases), type at the command prompt:
> denss.py -f 6lyz.outThe estimated maximum dimension, Dmax, is required for setting the size of the box. When possible, the Dmax is extracted from the file. This is possible for file types that store Dmax and that DENSS can parse, such as GNOM .out files and DENSS .fit files. If Dmax cannot be extracted from the file, DENSS can also automatically estimate Dmax. When using a standard .dat file containing raw data, DENSS will attempt to fit the data while estimating Dmax. In all cases, Dmax extracted from the file or estimated by DENSS can be overridden using the -d option as follows:
> denss.py -f 6lyz.out -d 50.0The box size DENSS creates is equal to Dmax*oversampling. Thus, this command tells DENSS to create a real space box with an edge length of 150 Å, since the default oversampling ratio is 3.
FAST, SLOW and MEMBRANE Modes
DENSS has convenient options for selecting some good defaults for the vast majority of cases. Three “modes” are now included as options to select these defaults, FAST mode, SLOW mode and MEMBRANE mode. The default mode is SLOW, since this is suitable for most cases and takes about 2-20 minutes or so on a modern processor*. If you have a relatively simple globular shape you can select FAST mode using the -m option of denss.py, which will take about 30 to 60 seconds*. If you have a membrane protein, or actually any object that might have negative contrast, you can use the MEMBRANE mode instead. Read below for more details on this mode.
*Note: When installing NumPy, if you install the latest versions with Anaconda, Numpy will often be installed with MKL. MKL allows multicore acceleration with NumPy. This provides a significant speed up for denss.py, particularly with processors with several cores.
SLOW mode will set the box size to be Dmax * oversampling, and will set the number of samples (N) to 64 by adjusting the voxel size. FAST mode will use 32 samples instead. Other options affected by these modes can be found on the Advanced Options page. All of the options can still be overridden when using these modes by explicitly setting them.
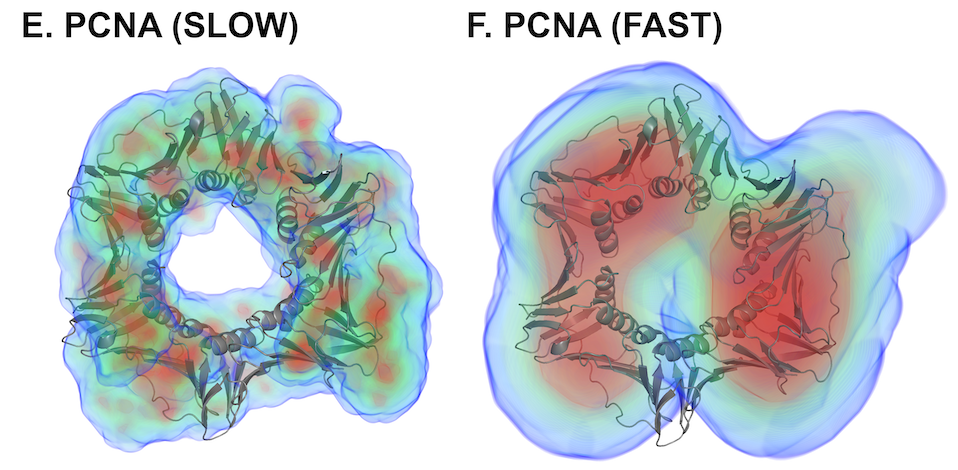
Here is an example of the difference between FAST mode and SLOW mode:
MEMBRANE mode, new as of version 1.4.9, disables the positivity restraint and thus allows for negative contrast. Similar to SLOW mode, MEMBRANE mode will use 64 samples. However, since positivity is turned off, shrink-wrap will start immediately. While this mode is designed for objects with negative contrast, it will work even in cases that do not have significant negative contrast. However, note that due to disabling the positivity restraint, density maps may be noisier than usual and occasionally require more reconstructions during the averaging step.
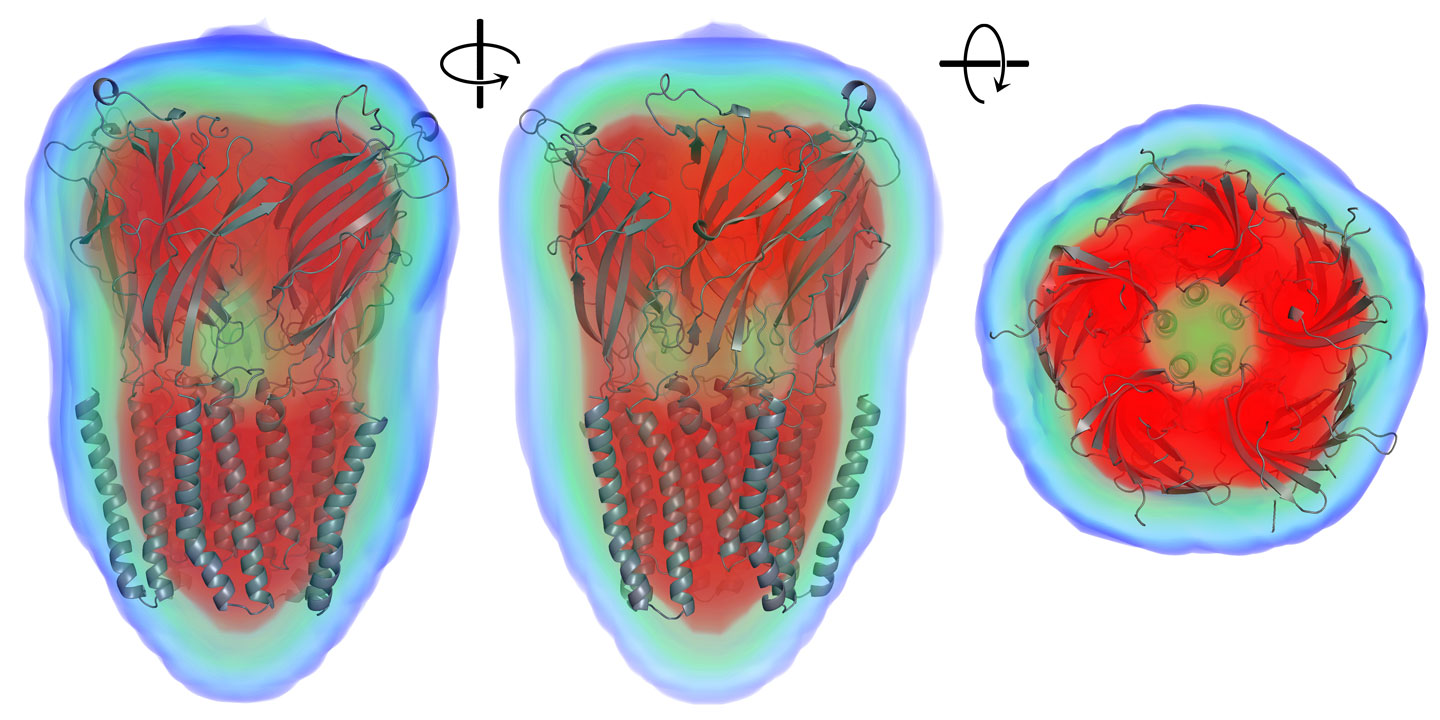
MEMBRANE mode may also be useful for SANS data, which often shows variable contrast for different components of a particle. Here is an example of a reconstruction of a membrane protein from SANS data using 5-fold symmetry (SASDL33):
Since N must be an integer, the voxel size is adjusted to the nearest value which yields an integer value for N, even if you set an explicit voxel size. You can check the .log file and look for the line saying “Real space voxel size” to find out what the actual voxel size used was, to be distinguished from the line saying “Requested real space voxel size” which will tell you what the voxel size input to the program through the -v option was.
Other commonly used options include:
| -f FILE, –file FILE | SAS data file for input (either .dat, .fit, or .out) |
| -u UNITS, –units UNITS | Angular units (“a” [1/angstrom] or “nm” [1/nanometer]; default=”a”) |
| -m MODE, –mode MODE | Mode. F(AST) sets default options to run quickly for simple particle shapes. S(LOW) useful for more complex molecules. M(EMBRANE) mode allows for negative contrast. (default SLOW) |
| -d DMAX, –dmax DMAX | Estimated maximum dimension |
| -n NSAMPLES, –nsamples NSAMPLES | Number of samples, i.e. grid points, along a single dimension. (Sets voxel size, overridden by –voxel. Best optimization with n=power of 2. Default=64) |
| -ncs NCS, –ncs NCS | Rotational symmetry |
| -ncs_steps NCS_STEPS [NCS_STEPS …], –ncs_steps NCS_STEPS [NCS_STEPS …] | Space separated list of steps for applying NCS averaging (default=3000 5000 7000 9000) |
| -ncs_axis NCS_AXIS, –ncs_axis NCS_AXIS | Rotational symmetry axis (options: 1, 2, or 3 corresponding to (long,medium,short) principal axes) |
| -ncs_type NCS_TYPE, –ncs_type NCS_TYPE | Symmetry type, either cyclical (default) or dihedral (i.e. C or D, dihedral (Dn) adds n 2-fold perpendicular axes) |
| -s STEPS, –steps STEPS | Maximum number of steps (iterations) |
| -v VOXEL, –voxel VOXEL | Set desired voxel size, setting resolution of map |
| -os OVERSAMPLING, –oversampling OVERSAMPLING | Sampling ratio |
| –ne NE | Number of electrons in object (default 10000, set to negative number to ignore.) |
| -cef CHI_END_FRACTION, –chi_end_fraction CHI_END_FRACTION | Convergence criterion. Minimum threshold of chi2 std dev, as a fraction of the median chi2 of last 100 steps. |
| -o OUTPUT, –output OUTPUT | Output filename prefix |
| -gpu, –gpu | Use GPU acceleration (requires CuPy). (default False) |
NCS Symmetry Averaging
Since v1.4.6, denss.py includes an option for utilizing symmetry averaging to improve the accuracy of reconstructions. The NCS (non-crystallography symmetry) averaging procedure works by first aligning the principal axes of the map to the x, y, z axes, sorted from longest to shortest axis. Then the symmetry mate for the given rotational symmetry operator (simple N-fold rotation about the principal axis) is calculated (via interpolation), and the density value at each rotational position is set to be the average of all density values at those positions. To invoke NCS symmetry averaging, simply set the –ncs option. For example, to impose 2-fold symmetry type at the command prompt:
> denss.py -f 6lyz.out -ncs 2Now for lysozyme this doesn’t make sense, since 6LYZ does not have 2-fold symmetry, but is just shown for illustration. By default the symmetry averaging will be performed a few times, at step 3000, 5000, 7000, 9000. To impose NCS at alternative steps, set the –ncs_steps option. This option takes a space-separated list of steps. Selecting more steps will lead to a stronger NCS restraint, but with the consequence of imposing more bias (and sometimes artifacts of interpolation, i.e. smearing or stripes). Since the NCS is not imposed at every step, the reconstruction is not forced to be symmetrical. Also by default the symmetry axis is assumed to be the longest principal axis. However, in many cases the longest axis is not the symmetry axis. To select a different symmetry axis, invoke the –ncs_axis option. This option takes an integer, either 1, 2, or 3. By default this is set to 1, i.e. the first, or longest, axis. For example, to impose 3-fold symmetry at steps 3000, 5000, and 7000 about the shortest principal axis:
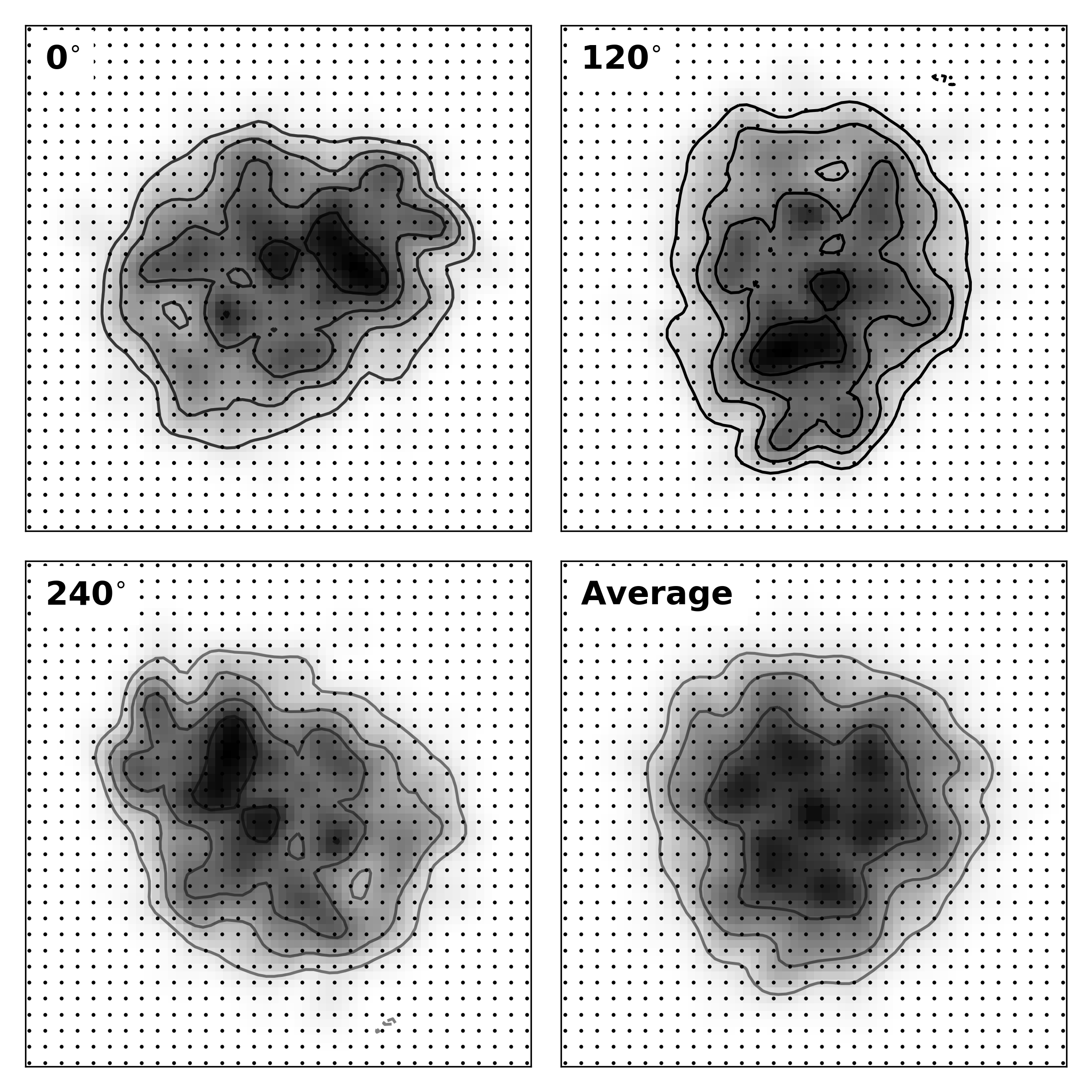
> denss.py -f 6lyz.out -ncs 3 -ncs_steps 3000 5000 7000 -ncs_axis 3Below is an image showing how the symmetry averaging is carried out. First, the principle axes of inertia are calculated and sorted from longest to shortest. Next, the density is recentered and reoriented such that the longest axis is along the X axis, the middle axis along Y, and the shortest axis along Z. Then, the density is rotated N times about the user-defined axis of symmetry by 360/N degrees to generate the symmetry mates. Then all of the symmetry mates are averaged together to generate the symmetry averaged density.
Dihedral symmetry:
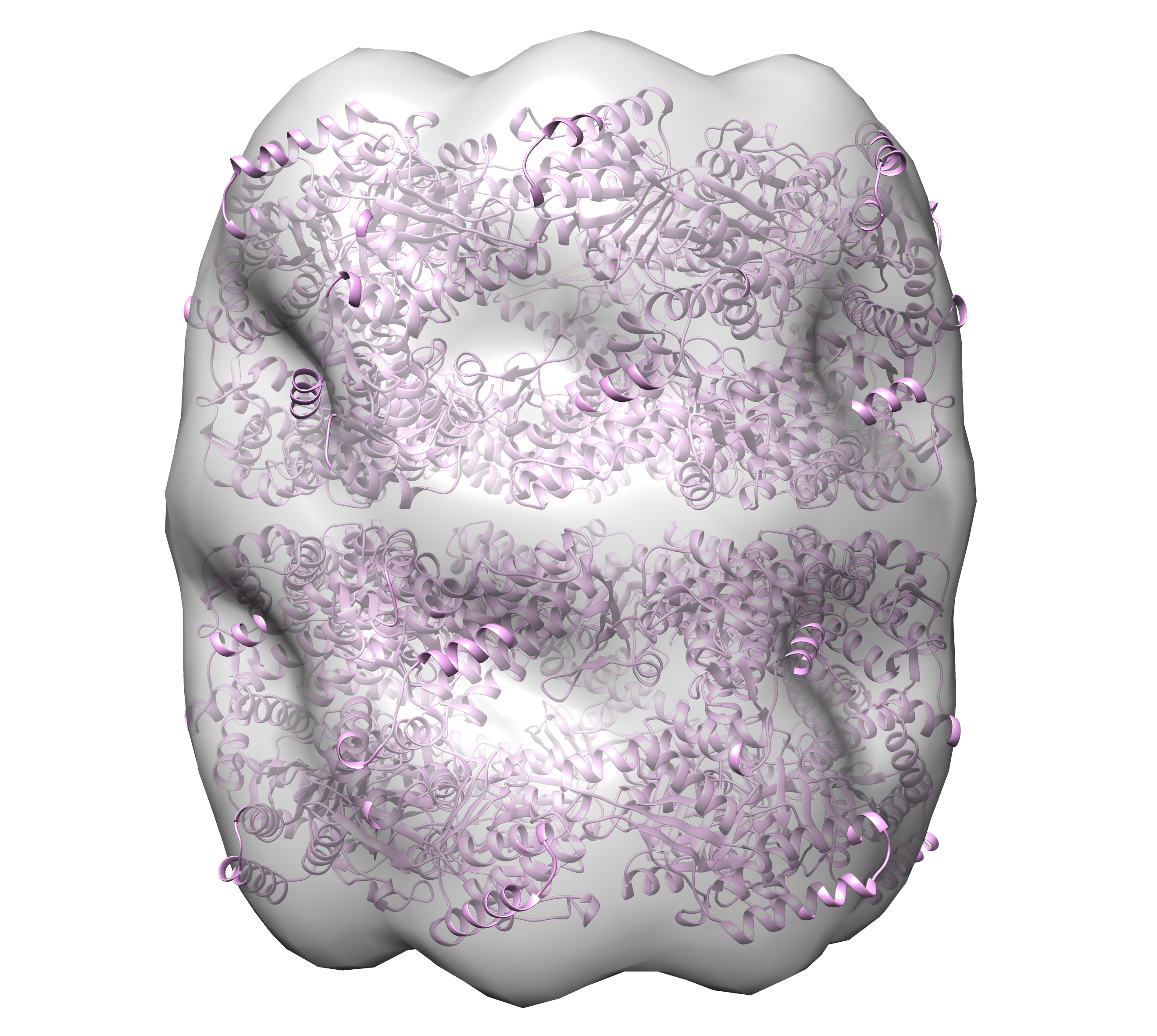
In addition to simple rotational symmetry (cyclical symmetry about a single axis), DENSS also has the ability to impose dihedral symmetry using the “–ncs_type dihedral” option. In this case, there is a single axis of rotation for the rotational symmetry, and then N additional rotational axes for the two-fold dihedral symmetry. For example, GroEL has 14 subunits arranged in D7 symmetry, so there are 7 dimers, where each dimer contains a two-fold axis perpendicular to the 7-fold axis of symmetry. In the case of GroEL the 7-fold axis of symmetry is along the longest principle axis. In this case the command to run DENSS with full averaging and D7 dihedral symmetry using 4 cores would be:
> denss.all.py -f groel_saxs.fit -ncs 7 -ncs_type dihedral -ncs_axis 1 -j 4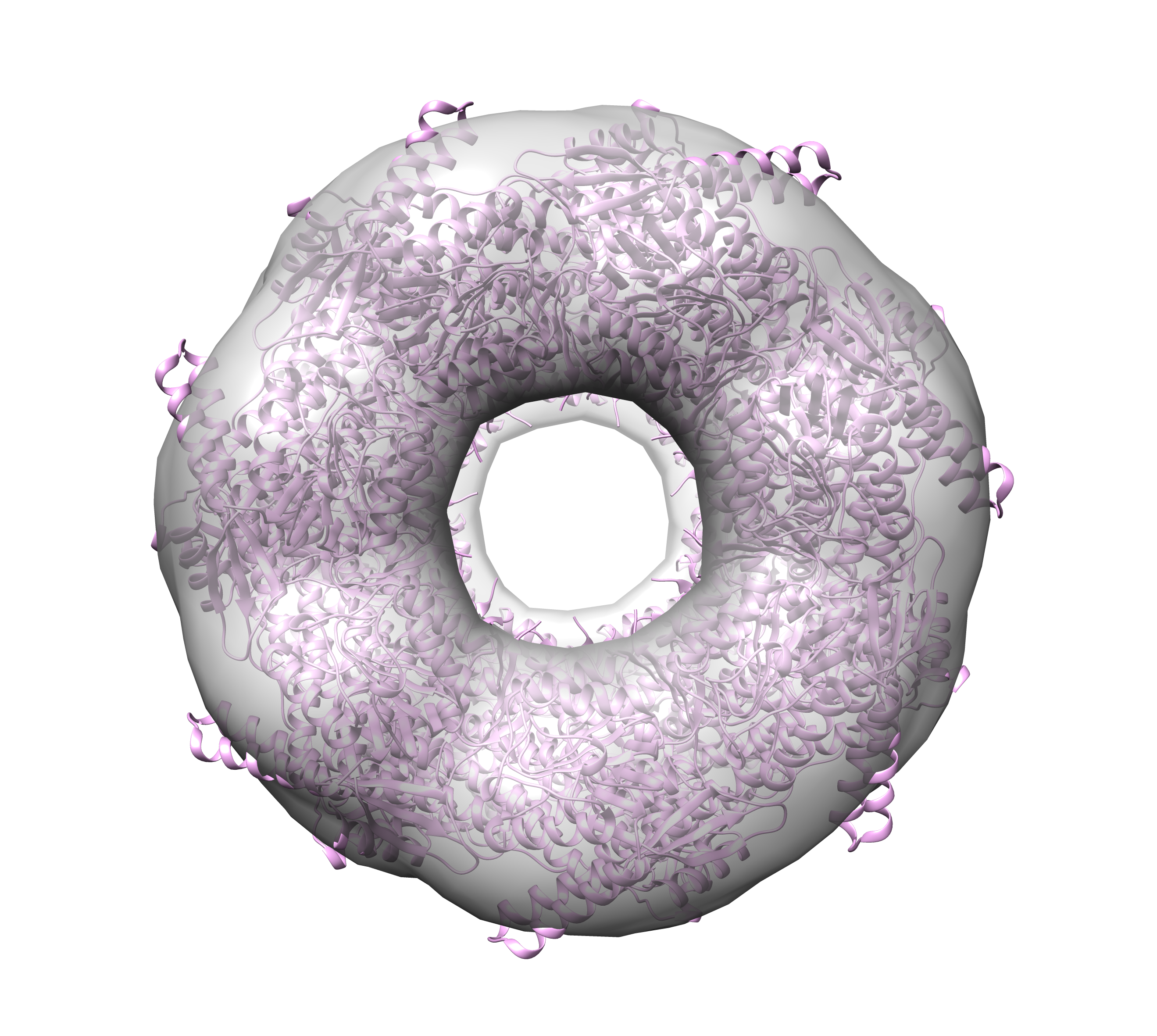
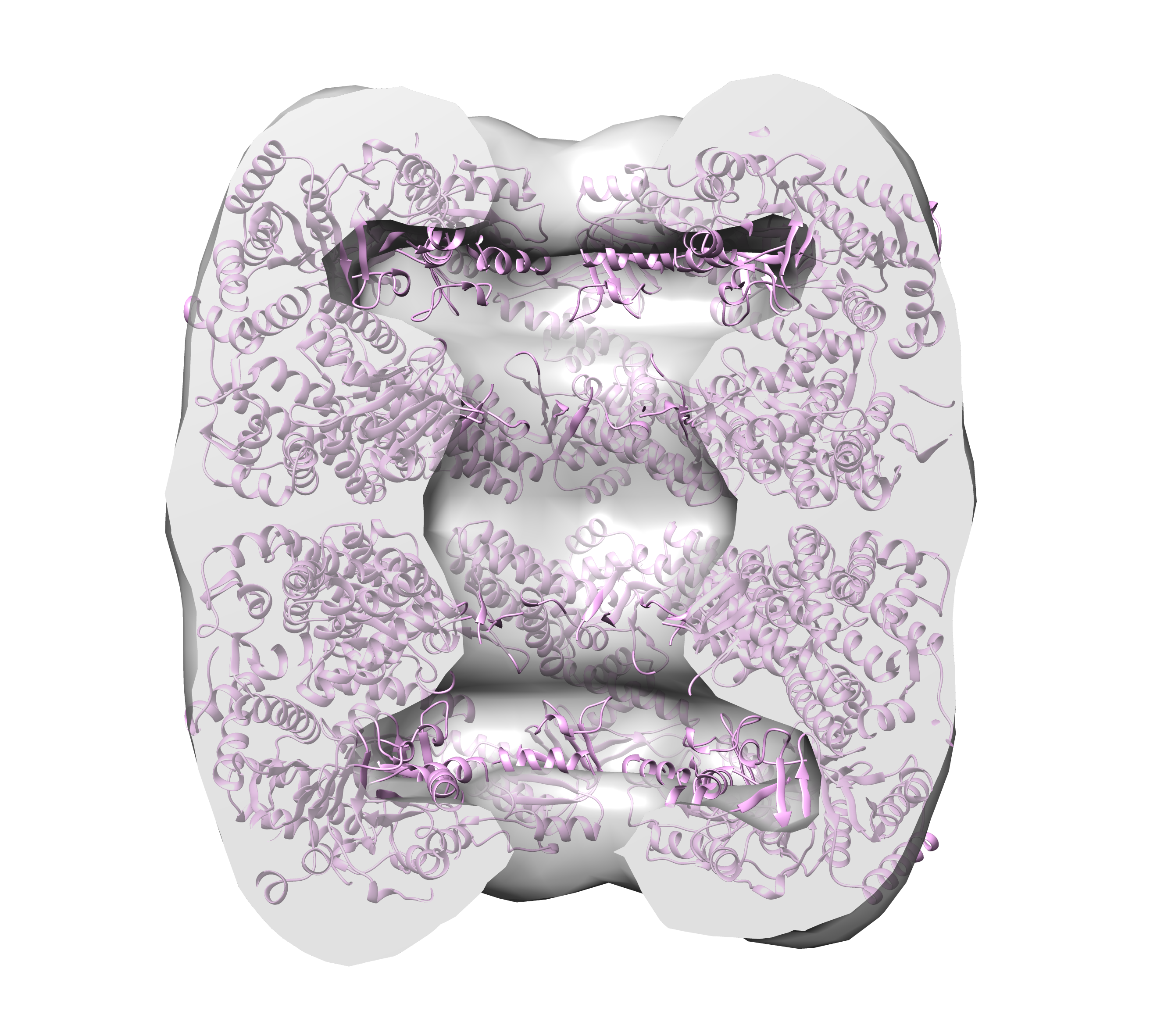
TIP: If you find that the output that DENSS produces does not show clear symmetry, it is possible that DENSS converged prior to the first step of applying symmetry, i.e. before step 3000. Check your log file to see how many steps DENSS ran before converging and stopping. If the total number of steps is less than the first step in ncs_steps, then DENSS never got a chance to apply symmetry. If this is the case, you can decrease the convergence criterion using the -cef option (default=0.001), or remove it entirely by setting it to zero.
Calculation Time
The speed of the program is pretty much entirely dictated by N. In some cases the algorithm will converge in fewer steps than in other cases, so it does vary some in that sense. For simple cases where N = 32, a single run of denss.py takes about 20 seconds or so on a modern processor (e.g. 2.5 GHz, Intel i5). For the default case where N = 64, it takes about 3 minutes; for N = 128 it takes over 30 minutes. However, as noted below, running multiple reconstructions (20 for simple cases, maybe 100 for complex cases) is required, and the averaging process tends to take longer than the reconstructions themselves.
Results
As the program runs, the current status will be printed to the screen like so:
> denss.py -f 6lyz.out
Step Chi2 Rg Support Volume
----- --------- ------- --------------
349 8.35e+04 34.76 3375000
These values will update in line as the program progresses. “Step” is the current iteration, “Chi2” (or rather χ2) is the goodness of fit of the calculated intensity versus the interpolated experimental intensity. “Rg” is the radius of gyration calculated directly from the electron density map. “Support Volume” is the total volume in Å3 of the support, i.e. the voxels containing the particle which is determined by shrink-wrap (see below).
Additionally, when the –enforce_connectivity restraint is imposed, an additional integer number will be printed to the right of the Support Volume and the results line will be kicked down one line. This number refers to the number of “features” that the –enforce_connectivity option counted, i.e. the number of separate blobs. If you see this number is 1, then that means shrinkwrap already got rid of all the other disconnected blobs of density and it won’t have much effect. This happens more often for simple globular particles. However, for less globular particle shapes, there are often several disconnected features at this early stage of the reconstruction, so the –enforce_connectivity restraint eliminates all of the minor features, retaining only the feature with the greatest density. It will look something like this:
> denss.py -f 6lyz.out
Step Chi2 Rg Support Volume
----- --------- ------- --------------
5999 5.62e+01 16.36 52234 1
8259 1.31e+00 14.34 42135
Some notes about these values:
In most cases, the χ2 value reported should only be used as a relative indicator of whether or not convergence is occurring. χ2 is very sensitive to accurate error estimates and can easily be orders of magnitude off in scale (particularly here as we are interpolating values and using smooth curves which do not adjust errors to account for oversampling). However as the reconstruction progresses, you should notice a steady decline in the reported value. Do not be concerned if the value fluctuates up and down (particularly around the step where the –enforce_connectivity option is invoked), as this is often part of the convergence process.
Due to the random nature of the starting seed of the algorithm, multiple reconstructions often vary in final Rg by a few (maybe 5 or so) percent.
The “Support Volume” is only the volume of the support region and should not be confused as the actual volume of the particle. The “Support Volume” will always be larger than the volume of the particle since it is the volume of the voxels containing the particle. However it should give you some idea of an upper bound of the particle volume. To actually estimate particle volume from the density, you can open up the .mrc file in Chimera and open the “Measure Volume and Area” tool. This will calculate the volume of the particle at the given density threshold (which is user defined).
When the program stops:
DENSS will stop when either one of the following two things happen: (1) the maximum number of steps has been reached (–steps option) or (2) when convergence has been achieved (-cef option). Here, “convergence” refers to when the chi2 stops changing. Convergence is defined as when the standard deviation of the chi2 over the previous 100 steps is less than some threshold, i.e., when the fluctuations or change in chi2 is small. The threshold is defined as a fraction of the median chi2 from that same previous 100 steps. By default, the threshold fraction (i.e, the -cef option) is set to 0.001. If you find that the program converges and stops earlier than you would like (for example if it stops before first imposing symmetry at step 3000), then you can set the -cef option to be a smaller number. If you would like to remove this convergence criterion completely, such that the program only stops when the maximum number of steps has been reached, then set the -cef option to zero.
Output Files
Electron density maps are written in CCP4/MRC format (credit Andrew Bruno) and optionally as Xplor ASCII text format (with the –write_xplor option enabled). These files can be opened directly in some visualization programs such as Chimera and PyMOL. In particular, the PyMOL “volume” function is well suited for displaying these maps with density information displayed as varying color and opacity. Maps can be converted to other formats using tools such as the Situs map2map tool or the EMAN2 e2proc3d.py program.
Output files for denss.py include:
| output.mrc | Electron density map (MRC format) |
| output_support.mrc | Final support volume formatted as unitary (0 or 1) electron density map |
| output_stats_by_step.dat | Statistics as a function of step number. Three columns: chi^2, Rg, support volume. |
| output_map.fit | The fit of the calculated scattering profile to the experimental data. Experimental data has been interpolated to the q values used for scaling intensities and I(0) has been scaled to the square of the number of electrons in the particle. Columns are: q(data), I(data), error(data), q(calc), I(calc) |
| output_*.png | If plotting is enabled, these are plots of the results. |
| output.log | A log file containing parameters for the calculation and summary of the results. |
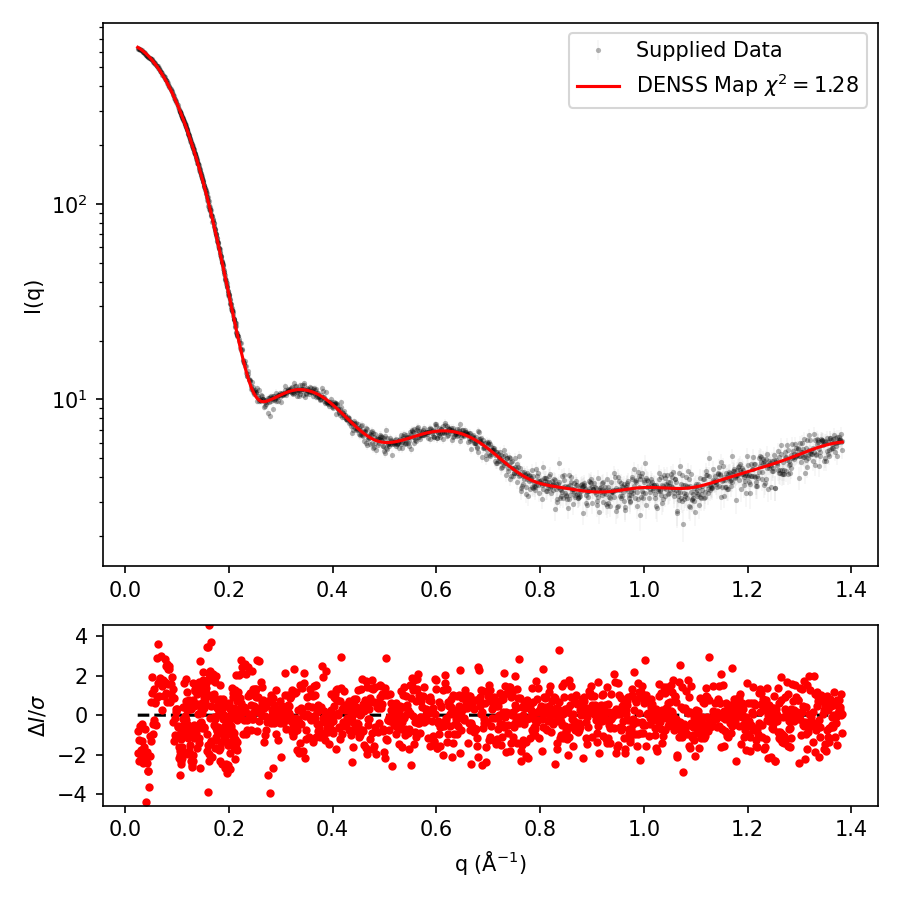
One of the key methods to assess whether or not DENSS worked is to check the output_fit.png file containing the plot of the fit of the DENSS reconstruction compared to the experimental data, including an actual chi2 value. An example of this plot is shown below. If this plot does not show good agreement, then you should discard the DENSS reconstruction, as it suggests it failed. In that case, try adjusting the parameters described above or in the Advanced Usage page (e.g., try increasing the oversampling to 5, try decreasing the shrink-wrap threshold option to 0.10 or 0.15, try MEMBRANE mode, etc.).