Tips
Most of the tips and best practices for reconstructing density using DENSS can be found throughout the Tutorial pages. Here I will just describe a few handy tips that aren’t mentioned elsewhere.
Evaluating the Results
The primary result from a single reconstruction of denss.py is the output.mrc file. This file contains the final electron density reconstructed by DENSS formatted as a CCP4/MRC file which is one of the most common volumetric data formats used. It can be opened by most volume visualization programs such as Chimera, PyMOL, EMAN2, and many others. After running the averaging procedure with denss.all.py or superdenss, the final averaged density can be found in the folder containing all of the output with the output prefix you specified with the -o option, named something like 6lyz_avg.mrc. Additionally, if you have the matplotlib module installed, a file called something like 6lyz_fsc.png will be created showing a plot of the Fourier Shell Correlation calculated from the averaging procedure with the estimated resolution given. The resolution is also printed to the terminal screen at the end of the averaging procedure.
Visualization
Chimera is an excellent tool for assessing many features about the electron density map. First open the 6lyz_avg.mrc file in Chimera and select an appropriate threshold (Tools > Volume Data > Volume Viewer). What’s an “appropriate” threshold? A good starting point would be the threshold that creates a contour of the density that encloses a total volume equal to the expected volume of the particle. For example, a rough estimate of protein volume (in Å3) is 1.7 * mass in kilodaltons. Alternatively, one can estimate the Porod volume using tools such as datporod in ATSAS or denss.fit_data.py, which will calculate volume also. To estimate the volume of the envelope contoured by the current threshold, open the Measure Volume and Area tool (Tools > Volume Data > Measure Volume and Area, make sure your volume object is selected in the drop down menu first). Then drag the threshold slider until you achieve the desired volume. Alternatively, you can automatically set the threshold based on your desired volume (in Å3) using the Chimera command line with the following command:
> volume #0 encloseVolume 17000where the #0 refers to the object ID (check the Tools > General Controls > Model Panel), and here the 17000 is the volume you want to enclose.
PyMOL is also great for visualizing the density. The “volume” representation is particularly useful for seeing the gradients of density inside the particle envelope. To load the .mrc file in PyMOL, open PyMOL and type at the PyMOL command prompt:
> load 6lyz.mrc
> volume 6lyz_volume, 6lyzThis will load your map and create a volume object from it. By default it will color the density as 2Fo – Fc, which in most cases will essentially give you an envelope of the particle. To see the internal density fluctuations we need to create a new color “ramp”. This can be done in one of two ways.
First, to color your density interactively, use the volume adjustments dialog by selecting the “C” (Coloring) box next to the object name, and select “panel”. In the new window that pops up, adjust the opacity of the dots by dragging them up or down or adjust the electron density value by dragging them right or left.
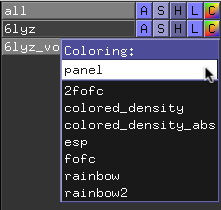
You can also explicitly create a color ramp on the PyMOL command line by typing:
> volume_ramp_new colored_density, 2 blue 0.0 2.5 blue 0.01 5 cyan 0.01 7.5 green 0.01 10 yellow 0.01 15 red 0.01 200 red 0.03If you’re using the new PyMOL 3, the density maps have different default normalization, so this should work:
> volume_ramp_new colored_density, 0.08 blue 0 0.10 blue 0.025 0.48 green 0 0.50 green 0.025 0.98 red 0 0.99 red 0.025 50 red 0.05This will create a new color ramp called “colored_density” that gradually changes color and opacity from transparent blue at the lowest densities through cyan, green, yellow and finally red at the highest densities. By default these values are given in units of standard electron density sigma from crystallography (rather than absolute e-/Å3) (unless you are using PyMOL 3). Note that DENSS reconstructions often have significant density at very high sigma values (much higher than crystallography) due to the fact that the box size is much larger than a typical crystallographic unit cell and the solvent region is filled with zeros.
Now that we have created the color ramp, let’s apply it to our volume object:
> volume_color 6lyz_volume, colored_densityand that should update the representation like this:


If you would like to make the new colored_density color ramp to be available every time you open PyMOL, add the volume_ramp_new line above to your .pymolrc file (likely in your $HOME directory, or create it if it doesn’t exist). Now you will be able to select the color ramp from the “C” box dropdown menu.
To adjust the values for viewing the density, open up the volume adjustments panel in PyMOL by selecting the “C” (Coloring) box next to the object name, and select “panel”. In the new window that pops up, adjust the opacity of the dots by dragging them up or down or adjust the electron density value by dragging them right or left.
Assessing the Quality of the Reconstructions
One of the first things to determine is whether the settings you have set for the individual runs of denss.py were sufficient for accurately reconstructing the particle. While the default values are suitable for most standard cases, for more complex cases, such as particles with multiple connected domains, or molecules with large aspect ratios (e.g. > 5:1), it is not uncommon for the fit of the scattering profile to be poor relative to the experimental data. This can be evaluated by viewing the output_fit.png file which contains a plot of the intensity versus q values for the q-shells used in the reconstruction. Note that after the last iteration of DENSS, the structure factors are scaled one final time to ensure that the intensities match exactly that from the input data. However, just prior to the final scaling of the structure factors, after the real space restraints are imposed for the last time, the fit of the calculated intensities versus the experimental intensities is calculated and recorded, and ultimately plotted in the output_fit.png file. This is to give an indication of the quality of the reconstruction, as by the end, after the process has converged, this plot should closely fit the experimental data.
Open the output_fit.png file. If you see that the red dots corresponding to the calculated intensities match well to the blue dots corresponding to the experimental intensities (interpolated at the required q values), then that suggests the reconstruction was successful. To get a precise assessment of this, the residuals between these curves is shown on the bottom of this plot:
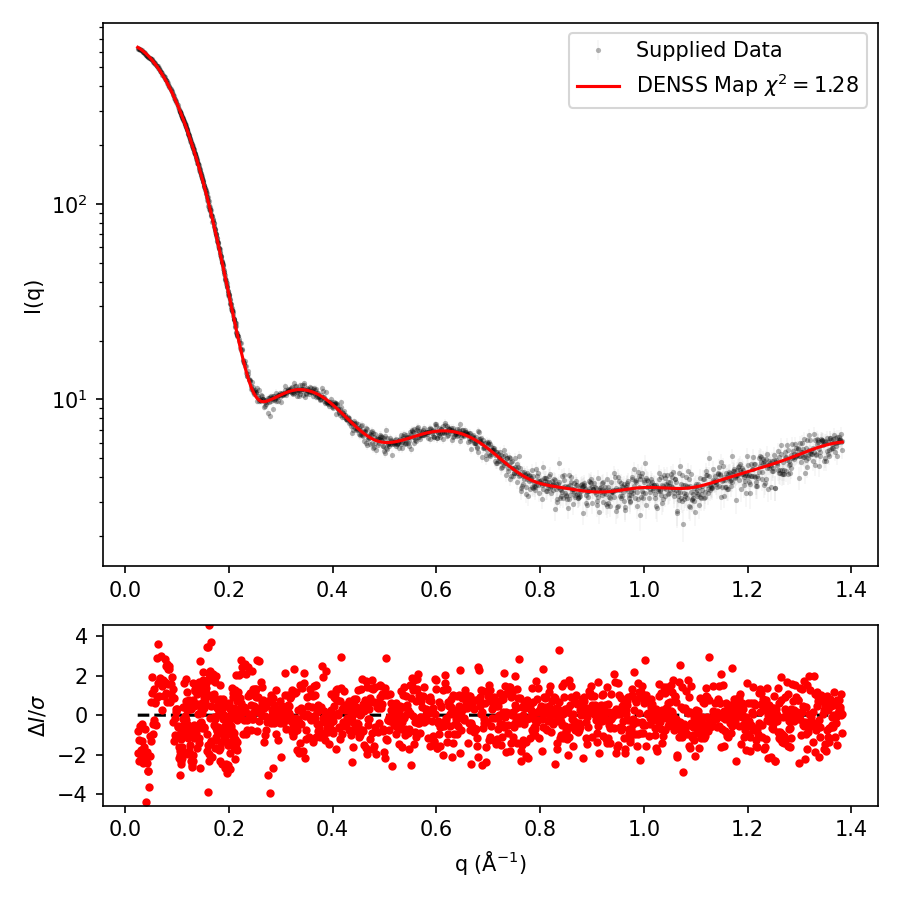
The above example shows a good fit between the two curves, suggesting that the reconstruction was successful. In some cases, e.g. complex shapes, the fit shown in this plot may be poor.
Things to try if DENSS fails (e.g., if the fit looks poor):
- Try increasing the oversampling ratio (-os option) to 5. This will increase the size of the box, improving the sampling of the scattering profile, particularly at low q values. Note that this will correspondingly increase the size of the real space voxel, so will reduce the resolution of the map in real space. If you would like to increase the oversampling and keep the voxel size the same, then increase the number of samples, -n. Note that this will take much longer to run (10-30 minutes). For example, to run with an oversampling of 6 and the same voxel size, run this command:
> denss.py -f saxs_data.fit -os 6 -n 128 - Try decreasing the shrink-wrap threshold (-sw_threshold) to 0.10 or 0.15. This will reduce the amount of clipping of the object that shrink-wrap causes, often allowing the object to be more elongated or complex in shape. Note that this will also allow more noise into the map, so you may need to average more reconstructions, such as 100 rather than the default 20, to get good quality averages.
- Try MEMBRANE mode. MEMBRANE mode disables the positivity restraint, which allows for negative contrast. Note that this can also result in good fits even if the particle is not well defined, so use with caution unless you know your particle has a lot of negative contrast. Do not combine this with a reduced shrink-wrap threshold mentioned above as there are often then too few restraints to ensure good quality reconstructions.
Assessing Convergence
Three additional plots saved in .png files (and the _stats_by_step.dat file) show how the χ2, Rg and Support Volume changed as the reconstruction progressed. Note that before the step when –enforce_connectivity removes extra regions of density in the solvent, these numbers are particularly inaccurate. The three plots for 6lyz.out using default parameters are shown below. The decay and ultimate plateau of χ2 indicates that the reconstruction has converged and additional steps will have little effect on the results.



Comparing Results with a Known Structure
Oftentimes you will want to compare the final electron density map with some known structure (or partially known structure), such as from crystallography. There are a few ways this can be done.
Alignment using DENSS
The built-in script denss.align.py can align the 6lyz_avg.mrc reconstruction to atomic coordinates 6lyz.pdb. This script will first calculate an electron density map from the pdb file, and then align the 6lyz_avg.mrc to the calculated map. One advantage of using this script is that enantiomer generation and selection is built in. While denss.all.py and the -e option of superdenss can generate and select the enantiomers that are most similar between multiple reconstructions from denss, there’s no guarantee that the selected enantiomer in the final average is actually the correct enantiomer, just that all the reconstructions represent a similar enantiomer for the purposes of averaging. Hence, if you would like to compare your final averaged density to a known structural model, its possible that denss may have selected the wrong enantiomer, making comparison with your structure difficult.
To perform this alignment with enantiomer selection (enabled by default), type:
> denss.align.py -f 6lyz_avg.mrc -ref 6lyz.pdbThis script will save three new files: 6lyz_avg_aligned.mrc (the aligned map); 6lyz_centered.pdb (the recentered pdb file); 6lyz_pdb.mrc (the map calculated from the recentered pdb file). You can then open these files in Chimera or PyMOL and see how they align.
If you would like to quantify how well the maps align, you can use the denss.calcfsc.py script. This script takes in two electron density maps (pre-aligned) and simply calculates the Fourier Shell Correlation and estimated resolution (using the 0.5 cutoff). To use this script, we can give it the aligned map and the calculated map saved by the denss.align.py script:
> denss.calcfsc.py -f 6lyz_avg_aligned.mrc -ref 6lyz_pdb.mrcThis will save a 6lyz_avg_aligned_fsc.dat file with the FSC curve and a 6lyz_avg_aligned_fsc.png file with the plot. It will also print the estimated resolution to the screen.
Alignment using Chimera and EMAN2
Alignment can be done conveniently in Chimera (though there are no options here for automatically selecting the enantiomer, but this can be done manually using the “vop zflip #0” command). First open your 6lyz_avg.mrc file and set an appropriate threshold (see above). Then open your pdb file. Next, we need to align the pdb model to the volume. To start, we first manually move the pdb model to approximately overlap with the volume. To do this, deselect the “A” checkbox (A for “active”) next to the 6lyz_avg.mrc volume object in the Model Panel. This ensures that when we move the pdb molecule in the next step, the volume object will not move along with it. Then, using the middle mouse button (see here for setting mouse preferences), translate the pdb model to overlap with your volume as shown below. To make the volume semitransparent, type in the Chimera command line:
> volume #0 transparency 0.5Then rotate (left mouse button) until you get a rough alignment of the pdb model to the volume. Then to perform a finer alignment, open up the Fit in Map tool (Tools > Volume Data > Fit in Map). Select the pdb model from the “Fit” drop down menu (probably will start with “select atoms” in drop down menu) and your 6lyz_avg.mrc volume file in the “in map” drop down menu (which will probably already be filled in for you). Then click the “Fit” button a few times to perform a more accurate fit.


To save the aligned pdb file, go to File > Save PDB… in Chimera.
One can also calculate a FSC curve between the pdb model and the volume now that they are aligned, but its quite a bit more complicated than with the new denss.align.py script. First, we must create a volume object from our pdb model. In the Fit in Map window, click the “Options” button. Click the check box next to “Use map simulated from atoms, resolution”. We need to enter an appropriate resolution in the box. An appropriate resolution is that determined by the DENSS averaging procedure (see here for details). Enter the resolution in the box and press Enter. This should create a new molmap object in the Model Panel (though you may not see anything in the main window). Click on the check box under “S” in the Model Panel to show the map in the main window. To calculate FSC, the new map must be resampled onto the same grid as the 6lyz_avg.mrc map. To do this, type in the Chimera command line (make sure your model IDs are correct; here my molmap model is #1.1 in the Model Panel and my 6lyz_avg.mrc file is #0):
> vop resample #1.1 onGrid #0 boundingGrid FalseNow we need to save that resampled map as an MRC file. Go to the Volume Viewer window and select File > Save map as… (make sure you have selected the new resampled map object in the Volume Viewer, in my case its now ID #2). This will create a new .mrc file that we can use to calculate the FSC curve (be sure to save it in the same directory as where your 6lyz_avg.mrc file is for the next step).
Now go to your terminal (not in Chimera) and cd into the directory where you saved the new resampled MRC file and the 6lyz_avg.mrc file. We can use the e2proc3d.py program from EMAN2 to calculate an FSC curve with the following command:
> e2proc3d.py --calcfsc=6lyz_ali_resampled.mrc 6lyz.mrc 6lyz_pdb2map_fsc.txtwhich will create a new file called 6lyz_pdb2map_fsc.txt containing our FSC curve comparing the averaged DENSS reconstruction and the aligned and resampled 6LYZ pdb model.
Generating Enantiomers
While denss.all.py and the -e option of superdenss can generate and select the enantiomers that are most similar between multiple reconstructions from denss, there’s no guarantee that the selected enantiomer in the final average is actually the correct enantiomer, just that all the reconstructions represent a similar enantiomer for the purposes of averaging. Hence, if you would like to compare your final averaged density to a known structural model, its possible that denss may have selected the wrong enantiomer, making comparison with your structure difficult.
To generate and select the best enantiomer, use the procedure described in the denss.align.py script above.